
Philip Rosenfield, Alex X. Lu, Ava P. Amini, Lorin Crawford, Kasia Z. Kedzierska
Single-cell foundation models are an exciting paradigm for biologists, as they may accelerate the understanding of complex cell data and reveal previously unknown biology. Single-cell foundation models are pre-trained on datasets of millions of single-cell gene expression measurements. Their adoption is growing; these foundation models are being integrated into cell atlases and bioinformatics code packages for turnkey downstream usage. However, the utility of these models depends on the extent to which they are learning meaningful biology from large-scale databases. Our recently published paper in Genome Biology (opens in new tab) suggests they still have a long way to go.
Have single-cell foundation models learned general biology concepts?
One of the most impactful tasks single-cell foundation models should be able to do is make predictions on new, unseen data on which they were not explicitly trained – what the field refers to as “zero-shot” deployment. Zero-shot performance is critical to biological discovery. For example, strong capabilities in zero-shot inference would enable the discovery of cell types from unlabeled data, without further training on the new, unlabeled data. We hypothesized that analyzing the zero-shot performance of single-cell foundation models could help assess their potential for biological discovery and diagnose whether these models are learning meaningful biology.
We evaluated two popular single-cell foundation models, Geneformer and scGPT*. These models were created to analyze single-cell gene expression data. They are pretrained on tens of millions of single-cell profiles, learning patterns in how genes are expressed across different cell types and conditions. While we chose to focus on Geneformer and scGPT because these were models that had code available at time of our work, other single-cell foundation models typically train using a similar formula.
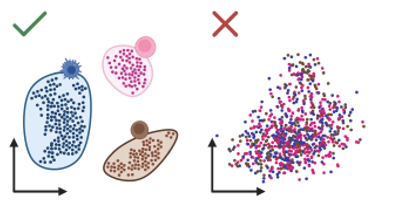
We assessed zero-shot performance in clustering cell types in five distinct datasets. In this task, the models must group together cells that perform the same function in tissue (e.g. acinar cells, which secrete digestive enzymes, versus endothelial cells, which line blood vessels). What makes this task challenging is that input data, for the same cell type, can look different depending on the experiment used to measure how genes are expressed in these cells, so models need to identify similarities between cells in the presence of these confounding technical effects or “batch effects”. A good model should cluster cells by cell type, and not by the type of experiment used for each cell (Figure 1).
Single-cell foundation models perform worse than traditional methods.
Across all datasets, Geneformer and scGPT perform poorly compared to more conventional machine learning methods like scVI, or statistical algorithms like Harmony (Figure 2). In some cases, they even perform worse than just using the highly variable genes (HVG) in a dataset, a basic feature selection strategy that uses the 2,000 most variable genes as opposed to all genes measured, or than an untrained version of the foundation models initialized to random weights.
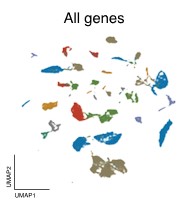




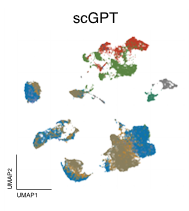
Left to right: no processing, traditional approaches (HVG, Harmony, scVI), single-cell foundation models (Geneformer, scGPT).
Why single-cell foundation models perform poorly
So, what could be the reason behind the observed poor performance of single-cell foundation models relative to simple, standard baselines? One explanation is that single-cell foundation models may not be learning to perform to the task that they are initially trained to do. These models are usually trained using masked gene expression prediction. During training, the model is shown input data with some genes withheld and is asked to predict the expression of these masked genes given the other genes. The logic behind this task is that models will have to learn relationships between genes – for example, if two genes are regulated by the same mechanism, or carry out the same function, they’re likely to be expressed in similar contexts.
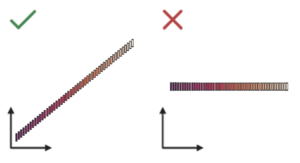
While this logic makes sense, we found that single-cell foundation models do not actually develop a deep understanding of this task. For example, we analyzed the ability of scGPT to predict the expression of held out genes. It is important to note that scGPT predicts gene expression in two ways: with and without utilizing the previously learned information stored within its cell embedding. In both cases, the model has limited ability to predict held out gene expression. Figure 3 shows what ideal versus poor results from gene expression prediction could look like. For the model to be considered high-performing, one would expect the bins to positively correlate and form a diagonal line rising in predicted expression (y-axis) with increasing input expression (x-axis; see Figure 3, left). Instead, without conditioning on the cell embedding (Figure 4, left), the model predicts the median expression value for every single gene regardless of its true expression value (see Figure 3, right). By conditioning on the cell embeddings (Figure 4, right), the ability of the model slightly improves, but only for the most highly expressed genes across datasets. Typically, these are “housekeeping” genes that are present at high levels of expression no matter what, so it’s questionable if single-cell foundation models, at least in their current form, learn the deeper relationships between genes that are variable depending on cell context.
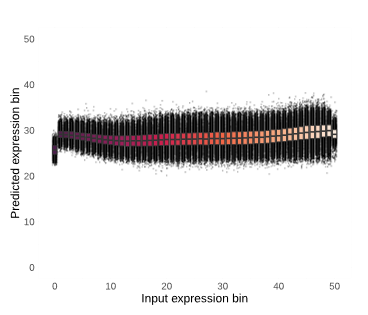
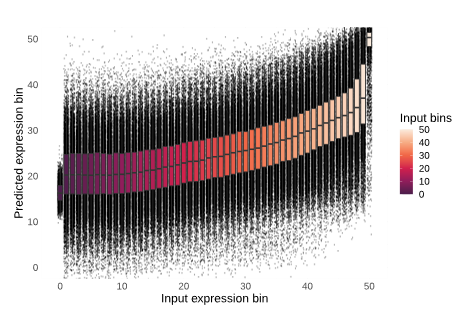
Overall, we demonstrate that single-cell foundation models perform poorly when deployed zero-shot. Despite this, there are cases where zero-shot embeddings from these models are already being integrated into cell atlases or bioinformatics analysis code. Our results caution against unprincipled adoption of current single-cell foundation models: we show that simpler baselines may outperform these recently proposed approaches, suggesting that practitioners should continue to consider using standard bioinformatic methods in practice.
Is there hope for improvement in single-cell foundation models?
Our results invite discussion on standards for evaluating single-cell foundation models. Prior to our work, the poor performance of these models in zero-shot settings went undetected, because most works showcased models in settings where they were further trained to specialize to specific downstream tasks, i.e., fine-tuned. However, fine-tuned evaluation set-ups can be vulnerable to misinterpretation, because increased performance on downstream tasks can be driven by statistical artifacts as opposed to learning meaningful biology. We previously exposed this trend for protein language models (opens in new tab), finding that bigger models with more parameters can sometimes capture task-relevant information by random chance versus capturing the underlying biology. Our observations across single-cell and protein models show that evaluation practices are still emerging for biological foundation models, and that thinking critically about how evaluations are setup and executed is necessary to develop AI models that learn meaningful representations of biology.
*For Geneformer we used the 6L architecture and for scGPT we show figures for the model trained on all cell types from their CellxGene dataset. Please see our paper (opens in new tab) for more details.

